利用凝胶层析介质(固定相)交联度的不同所形成的网状孔径的大小,在层析时能阻止比网孔直径大的生物大分子通过。利用流动相中溶质的分子量大小差异而进行分离的一种方法,称之为排阻层析。如下图所示
利用固定相载体上偶联的亚胺基乙二酸为配基与二价金属离子发生螯合作用,结合在固定相上,二价金属离子可以与流动相中含有的半胱氨酸、组氨酸、咪唑及其类似物发生特异螯合作用而进行分离的方法,称之为金属螯合层析。如下图所示
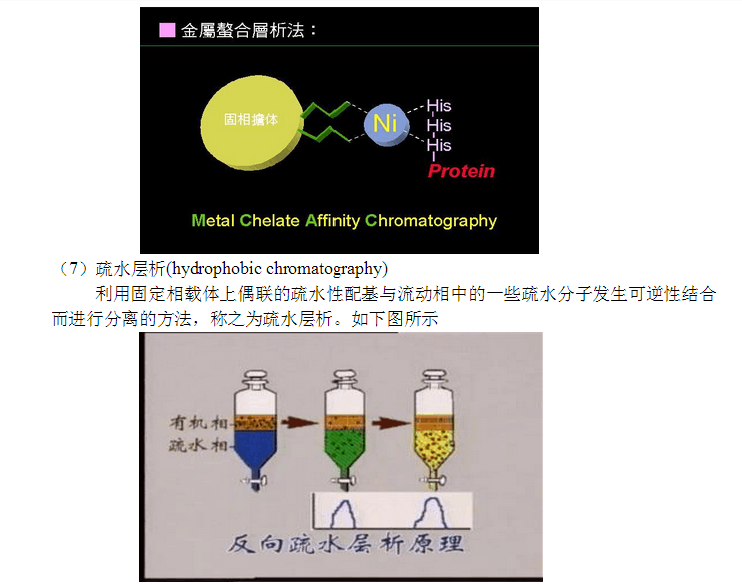
(8)反向层析(reverse phase chromatography)
利用固定相载体上偶联的疏水性较强的配基,在一定非极性的溶剂中能够与溶剂中的疏水分子发生作用,以非极性配基为固定相,极性溶剂为流动相来分离不同极性的物质的方法,称之为反相层析。
(9)聚焦层析(focusing chromatography)
利用固定相载体上偶联的载体两性电解质分子,在层析过程中所形成的pH梯度,并与流动相中不同等电点的分子发生聚焦反应进行分离的方法,称之为聚焦层析。 (10)灌注层析(perfusion chromatography)
利用刚性较强的层析介质颗粒中具有的不同大小贯穿孔与流动相中溶质分子分子量的差异进行分离的方法,称之为灌注层析。 2. 常用层析技术
2.1 离子交换层析技术
是以离子交换纤维素或以离子交换葡聚糖凝胶为固定相,以蛋白质等样品为移动相,分离和提纯蛋白质、核酸、酶、激素和多糖等的一项技术。 2.1.1 原理
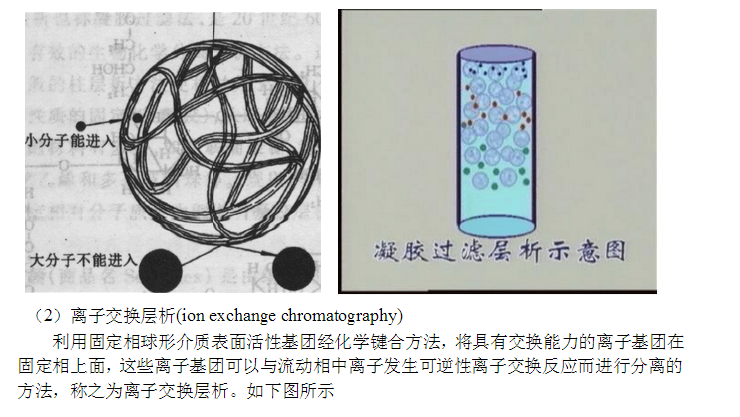
在纤维素与葡聚糖分子上结合有一定的离子基团,当结合阳离子基团时,可换出阴离子,则称为阴离子交换剂。如二乙氨乙基(Dicthylaminoethyl,DEAE)纤维素。在纤维素上结合了DEAE,含有带正电荷的阳离子纤维素—O—C6 H14N+H,它的反离子为阴离子(如Cl-等),可与带负电荷的蛋白质阴离子进行交换。当结合阴离子基团时,可置换阳离子,称为阳离子交换剂,如羧甲基(Carboxymethy, CM)纤维素。纤维素分子上带有负电荷的阴离
子(纤维素-O-CH2-COO一
),其反离子为阳离子(如Na+等),可与带正电荷蛋白质阳离子进行交换。
溶液的pH值与蛋白质等电点相同时,静电荷为0,当溶液pH值大于蛋白质等电点时,则羧基游离,蛋白质带负电荷。反之,溶液的pH值小于蛋白质等电点时,则氨基电离,蛋白质带正电荷。溶液的pH值距蛋白质等电点越远,蛋白质的电荷越多。反之则越少。血清蛋白质均带负电荷,但各种蛋白质带负电荷的程度有所差异,以白蛋白为**多,依次为球 蛋白,β球蛋白和γ球蛋白
在适当的盐浓度下,溶液的pH值高于等电点时,蛋白质被阴离子交换剂所吸附;当溶液的pH值低于等电点时,蛋白质被阳离子交换剂所吸附。由于各种蛋白质所带的电荷不同。它们与交换剂的结合程度也不同,只要溶液pH值发生改变,就会直接影响到蛋白质与交换剂的吸附,从而可能把不同的蛋白质逐个分离开来。
交换剂对胶体离子(如蛋白质)和无机盐离子(如NaCl)都具有交换吸附的能力,当#p#分页标题#e#
两者同时存在于一个层析过程中,则产生竞争性的交换吸附。当Cl一
的浓度大时,蛋白质不
容易被吸附,吸附后也易于被洗脱,当Cl一
浓度小时,蛋白质易被吸附,吸附后也不容易被洗脱。因此,在离子交换层析中,一般采用两种方法达到分离蛋白质的目的。一种是增加洗脱液的离子强度,一种是改变洗脱液的pH值。pH值增高时,抑制蛋白质阳离子化,随之对阳离子交换剂的吸附力减弱。pH值降低时,抑制蛋白质阴离子化,随之降低了蛋白质对阴离子交换剂的吸附。当使用阴离子交换剂时,增加盐离子,则降低pH值。当使用阳离子交换剂时,增加盐离子浓度,则升高溶液pH值。 2.1.2常用离子交换剂的种类与特性
(1)离子交换纤维素 离子交换纤维素的种类很多,**常用的是DEAE—纤维素和CM纤维素。由于剂型不同,其理化性质和作用也有所差异。一般而言,微粒型要优于纤维素型,因为微粒型是在纤维素型的基础上进一步提炼而成。它的交换容量大,粒细、比重大,能装成紧密的层析柱,要求分辨力高的实验可用此型纤维素。
离子交换纤维素的优点为:①离子交换纤维素为开放性长链,具有较大的表面积,吸附容量**大;②离子基团少,排列稀疏,与蛋白质结合不太牢固,易于洗脱;③具有良好的稳定性,洗脱剂的选择范围广。
(2)离子交换交联葡聚糖 离子交换交联葡聚糖也是广泛使用的离子交换剂,它与离子交换纤维素不同点是载体不同。
离子交换交联葡聚糖有如下优点:①不会引起被分离物质的变性或失活;②非特异性吸附少;③交换容量大。
离子交换葡聚糖的选用,一般根据蛋白质的分子量而定。中等分子量(30 000-200 000)一般选A50和C50,而低分子量(<30 000和高分子量>200 000)均宜选用A25和C25。 2.1.3 试验方法
阴离子交换剂与阳离子交换剂的装柱和层析过程基本相同。交联葡聚糖的预处理只需充分溶胀和平衡,不需要除去细粒碎片和酸碱处理。其他步骤也基本同离子交换纤维素。
(1)剂型的选择 根据蛋白质在所用缓冲液pH值下带电荷的种类选择,如pH高于蛋白质等电点,应选阴离子交换剂,反之应选阳离子交换剂。一般情况下,DEAE-纤维素用于分离酸性蛋白,而CM纤维素用于分离碱性蛋白质。 下面以DEAE-纤维素操作为例,介绍试验方法
(2)膨胀活化 此步的目的在于除去杂质,暴露DEAE-纤维素上的极性基团。DEAE-纤维素的用量则根据柱容积的大小和所需过柱样品的量来决定。一般是1.0g DEAE-纤维素相当于6ml~8ml柱床体积。
称取所需的量,撒于0.5 mol/L NaOH溶液中(1g DEAE—纤维素干粉约需15倍NaOH液),浸泡1h左右,不时搅拌。抽滤(以布氏漏斗加两层滤纸或尼龙纱布抽滤),以蒸馏水洗涤,再抽滤,直**滤液近中性为止,再将纤维素浸泡于0.5Mol/L HCl中1h,同样抽滤液**近中性。再将纤维素浸于0.5 mol/L NaOH液中,同样处理,洗**中性。
(3)平衡 将DEAE—纤维素放入0.0l mol/L pH 7. 4 PB液中(即起始缓冲液),静止1 h,不时搅拌,待纤维素下沉后,倾去上清液或抽滤除去洗液,如此反复几次**倾出液体的pH值与加入的PB液的pH值相近时为止。
(4)装柱 层析柱的选择要大小、长度适当。一般而言,柱长和柱直径之比为10︰1~20︰1,柱的内径上下要均匀一致。用前将层析柱在清洁液内浸泡处理24 h,然后依次用常水、蒸馏水、起始缓冲液充分洗涤。
(5)上样 要层析的样品shou先必须用起始缓冲液(4℃)平衡过夜,中间可换液数次。将柱的上端打开,用吸管将纤维素柱上面的缓冲液吸出,不要吸净,留一薄层液面,以免空气进入。沿管壁缓缓加入样品,注意不要打乱纤维素表层。拧开下端的螺旋夹,使样品进入交换剂中,快要进完时,加1 ml~2 ml缓冲液冲洗柱壁,随即用多量的洗脱液洗脱。
样品的加量与DEAE—纤维素有一个**适比的关系,超过这个比值,吸附就不完全,直接影响到分离的纯度。经过粗提的—球蛋白50 mg~100 mg,用干重约4 g DEAE-纤维素装柱分离,可获得理想结果。
(6)洗脱 对于阴离子交换剂而言,洗脱的办法是使pH逐渐降低,而离子浓度逐渐升高。一般的办法,是稳定一个因素而改变另一个因素洗脱。洗脱可采用分段洗脱和连续洗脱法,前者较实用,后者较准确。
(7)洗脱液的收集 利用自动分步收集器收集,并以20%磺基水杨酸测试,当蛋白液下来时,开始分管收集,**无蛋白液为止。
(8)交换柱的再生 将使用过的DEAE-纤维素移入烧杯中,用2 mol/L NaCl液浸泡,抽滤并洗涤数次。如不立即使用,可加1/10 000的叠氮钠防腐,保存于4℃冰箱中。使用时,再以碱-酸-碱处理。
2.2 凝胶层析
凝胶层析又称为分子筛层析或凝胶过滤。具有分子筛作用的物质很多,如浮石、琼脂、琼脂糖、聚乙烯醇、聚丙烯酰胺、葡聚糖凝胶等。以葡聚糖凝胶应用**广,商品名是sephadex型号很多,从G10到G200,它的主要应用范围是:①分级分离各种抗原与抗体;②去掉复合物中的小分子物质。如除盐、荧光素和游离的放射性同位素以及水解的蛋白质碎片;③分析血清中的免疫复合物;④分子量的测定。 2.2.1 原理 #p#分页标题#e#
凝胶是一种多孔性的不带表面电荷的物质,当带有多种成分的样品溶液在凝胶内运动时,由于它们的分子量不同而表现出速度的快慢,在缓冲液洗脱时,分子量大的物质不能进入凝胶孔内,而在凝胶间几乎是垂直的向下运动,而分子量小的物质则进入凝胶孔内进行“绕道”运行,这样就可以按分子量的大小,先后流出凝胶柱,达到分离的目的。 2.2.2 葡聚糖凝胶的种类与性能 葡聚糖又名右旋糖酐,在它们的长链间以三氯环氧丙烷交联剂交联而成。葡聚糖凝胶具有很强的吸水性,交联度大,吸水性小,相反交联度小,吸水性大。商品名以SephadexG表示,G值越小,交联度越大,吸水性越小,G值越大,交联度越小,吸水性就越大,二者呈反比关系,G值大约为吸水量的10倍。由此可以根据床体积而估算出葡聚糖凝胶干粉的用量。
G25、G50有四种颗粒型号:粗(100µ~300µ)、中(50µ~150µ)、细(20µ~80µ)和超细(10µ~40µ)。G75~G200又有两种颗粒型号:中(40µ~120µ),超细(10µ~40µ)。颗粒越细,流速越慢,分离效果越好。 2.2.3 试验方法
(1)凝胶的选择 根据层析物质分子量的大小选择不同型号的凝胶,如除盐和除游离的荧光素,则可选用粗、中粒度的G28或G500,G250多用于分离蛋白质单体,G200多用于分离蛋白质凝胶聚合体等。
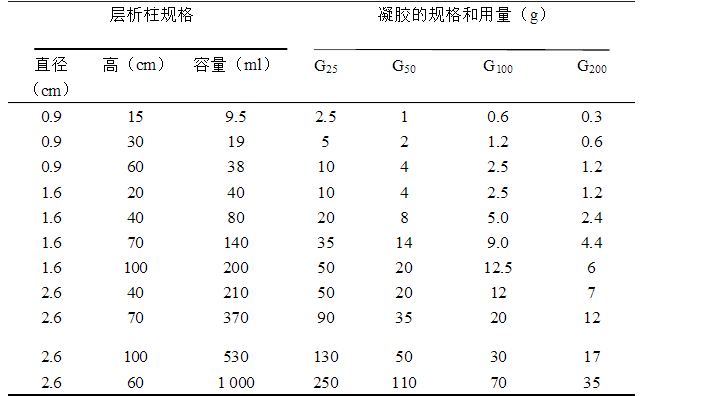
(2)凝胶的预处理 称取适量的凝胶加入过量的缓冲液在冰箱(或室温)中充分膨胀,或在沸水中煮,膨胀时间应根据不同型号的凝胶而定。如下表所示
凝胶量与型号和层析柱大小与规格及凝胶用量
为使粒子均匀一致需进行浮选,即加入凝胶粒子后,轻轻搅拌,静置20 min,倾去沉淀的粒子,如此反复数次即可。
(3)装柱 层析柱的选择一般根据分离样品的种类和样品的数量而定。纯化蛋白质时,柱床体积应为样品体积的25~100倍。去盐、游离荧光素约为样品体积的4~10倍。柱太短,影响分离效果。柱长一些,分离效果好,但柱太长,则延长分离时间,样品也稀释过度。层析柱的内径也要选择适当。内径过细,会发生“器壁效应”,即靠近管壁的流速要大于中心的流速影响分离效果。所以层析柱的内径和高度应有一定的比例。对于除盐来说应为1︰5~1︰25;对于纯化蛋白质来说应为1︰20~1︰100。 装柱过程基本同离子交换层析柱。
(4)加样与洗脱 样品体积不宜过多,**好为床体积的1%~5%,**多不要超过10%。样品浓度也不宜过大,浓度过大粘度大,分离效果差,一般不超过4%。
洗脱液应与膨胀一致,否则更换溶剂,凝胶体积会发生变化,影响分离效果。洗脱液要有一定的离子强度和pH值。分离血清蛋白常用0.02~0.1 mol/L pH 6.9~8.0的PBS液(0.14 mol/L NaCl)和0.1 mol/L pH8.0 Tris-HCl缓冲盐溶液(0.14 mol/L NaCl)。
(5)洗脱液收集 同离子交换层析。
(6)凝胶柱的重复使用与保存 当样品的各组分全部洗脱下来之后,即可加入新的样品,继续使用。保存方法有三种:
A 在液相中保存**方便,即于凝胶悬液中加入防腐剂(一般为0.02% N2N3或0.002%洗必泰)或高压灭菌后4℃保存。此法**少可以保存半年以上。
B 用完后,以水冲洗,然后用60%~70%酒精液冲洗,凝胶体积缩小,即在半收缩状态下保存。
C 长期不用者,**好以干燥状态保存,即水洗净后,用含乙醇的水洗,逐渐加大乙醇用量,**后用95%的乙醇洗,可全部去水,再用乙烯去除乙醇,抽滤干,于60℃~80℃干燥后保存。
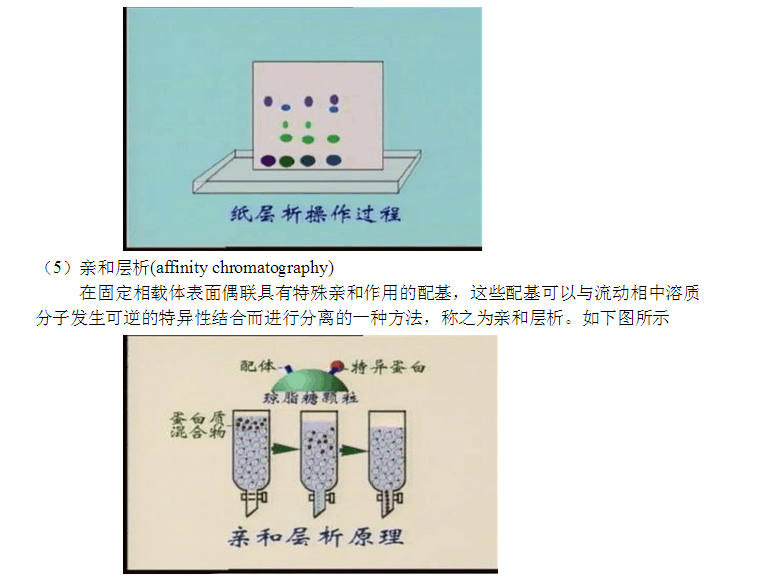
2.3 亲和层析 2.3.1 原理
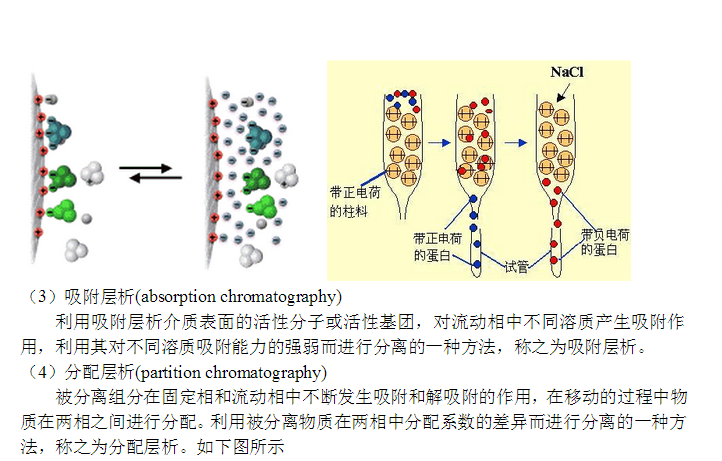
亲和层析是一种吸附层析,抗原(或抗体)和相应的抗体(或抗原)发生特异性结合,而这种结合在一定的条件下又是可逆的。所以将抗原(或抗体)固相化后,就可以使存在液相中的相应抗体(或抗原)选择性地结合在固相载体上,借以与液相中的其他蛋白质分开,达到分离提纯的目的。
此法具有高效、快速、简便等优点。 2.3.2载体的基本要求和选择
理想的载体应具有下列基本条件:①不溶于水,但高度亲水;②惰性物质,非特异性吸附少;③具有相当量的化学基团可供活化;④理化性质稳定;⑤机械性能好,具有一定的颗粒形式以保持一定的流速;⑥通透性好,**好为多孔的网状结构,使大分子能自由通过;⑦能抵抗微生物和醇的作用。
可以做为固相载体的有皂土、玻璃微球、石英微球、羟磷酸钙、氧化铝、聚丙烯酰胺凝胶、淀粉凝胶、葡聚糖凝胶、纤维素和琼脂糖。在这些载体中,皂土、玻璃微球等吸附能力弱,且不能防止非特异性吸附。纤维素的非特异性吸附强。聚丙稀酰胺凝胶是目前的**优良载体。
琼脂糖凝胶的优点是亲水性强,理化性质稳定,不受细菌和酶的作用,具有疏松的网状结构,在缓冲液离子浓度大于0.05 mol/L时,对蛋白质几乎没有非特异性吸附。琼脂糖凝胶极易被溴化氢活化,活化后性质稳定,能经受层析的各种条件,如0.1 mol/L NaOH或1 mol/L HCl处理2 h~3 h及蛋白质变性剂7 mol/L尿素或6 mol/L盐酸胍处理,不引起性质改变,故易于再生和反复使用。 #p#分页标题#e#
琼脂糖凝胶微球的商品名为Sepharose,含糖浓度为2%、4%、6%时分别称为2B、4B、6B。因为Sepharose 4B的结构比6B疏松,而吸附容量比2B大,所以4B应用**广。 2.3.3 试剂与配制 1.Sepharose 4B 2.CNBr(剧毒) 3.抗原或抗体
4.1 mol/L NaHCO3 取NaHCO3 84.01g加水**1 000 ml。 5.0.1 mol/L NaHCO3 6.2 mol/L NaOH
7.0.01 mol/L pH7.4 PBS
0.2 mol/L Na2HPO4 38.0 ml 0.2 mol/L Na2HPO4 162.0 ml NaCl 32.76 g 加水**4 000 ml。 8.0.1 mol/L pH 2.8 甘氨酸即氨基乙酸—HCl缓冲液 甘氨酸15.01 g加水**1 000 ml,取此液,加0.2 mol/L HCl 84 ml加水166 ml共500 ml。 9.7 mol/L尿素
10.0.2 mol/L NaHCO3,(含0.1 mol/L NaCI) 1 mol/L NaHCO3 100.00 ml NaCl 2.93 g 加水**500.00 ml。 2.3.4 实验方法 1.琼脂糖活化 ⑴ 取20 ml Sepharose 4B放在布氏漏斗中抽干,加少量的0.1 mol/L pH 9.0 NaHCO3液洗涤,立即转入100 ml烧杯中,冰浴置于磁力搅拌器上。 ⑵ 在通风橱内称取2 g溴化氰,加水20 ml溶解,然后倒入琼脂糖中,小心滴加2 mol/L NaOH,使pH保持在11左右,反应10 min。在1 min~2 min迅速调整pH为8.0~11.0维持10 min。 ⑶ 将活化的琼脂糖迅速倒入布氏漏斗中,以冰水抽洗成中性,再迅速以250 ml冷的0.1 mol/L pH 9.0 NaHCO3抽洗。
2.偶联蛋白 20 ml 4B液体相当于一半的固相载体。 ⑴ 事先将需偶联的抗原(或抗体)蛋白200 mg置于0.1 mol/L pH 9.0 NaHCO3液中透析数小时(一般偶联量为10~30 mg/g载体)。
⑵ 将活化的琼脂糖迅速倒入蛋白液中(在1.5 min内,从抽洗到偶联),4℃缓慢搅拌过夜,使蛋白与活化的琼脂结合。
3.装柱 ⑴ 选柱:不宜过大,一般以1.5×15 cm的层析柱可装偶联蛋白的琼脂糖约30ml。 ⑵ 装柱:将已与蛋白偶联的琼脂糖装入层析柱内,拧紧下口夹,让其下沉,数分钟后松开下口夹,使溶液约以1 ml/min的速度流出。 ⑶ 洗柱:以0.2 mol/L pH 9.0 NaHCO3(含0.1 mol/L NaCl)洗涤**洗出液的OD280<0.02为止。
收集全部的洗脱液,测得的OD280值×洗脱液的总ml数即为未偶联蛋白的含量,由此可计算偶联率。


 #p#分页标题#e#
#p#分页标题#e#


